Modifies ref sequence to personal sequence given the vcf
Show the code
def vcf_to_seq_faster(target_interval, individual, vcf_file, fasta_extractor):# target_inverval is a kipoiseq interval, e.g. kipoiseq.Interval("chr22", 18118779, 18145669)# individual is the id of the individual in the vcf file target_fa = fasta_extractor.extract(target_interval.resize(SEQUENCE_LENGTH)) window_coords = target_interval.resize(SEQUENCE_LENGTH)# two haplotypes per individual haplo_1 =list(target_fa[:]) haplo_2 =list(target_fa[:])# Open the VCF file vcf_reader = cyvcf2.VCF(vcf_file)# Specific genomic region CHROM = window_coords.chrom START =max(1,window_coords.start) END =min(window_coords.end, fasta_extractor._chromosome_sizes[CHROM]-1) count1 =0 count2 =0# Iterate through variants in the specified regionfor variant in vcf_reader(CHROM +':'+str(START) +'-'+str(END)):# Access the individual's genotype using index (0-based) of the sample individual_index = vcf_reader.samples.index(individual) genotype = variant.genotypes[individual_index] ALT=variant.ALT[0] REF=variant.REF POS=variant.POSif REF == target_fa[POS - window_coords.start -1]:if genotype[0] ==1: haplo_1[POS - window_coords.start -1] = ALT count1 = count1 +1if genotype[1] ==1: haplo_2[POS - window_coords.start -1] = ALT count2 = count2 +1else:print("ERROR: REF in vcf "+ REF +"!= REF from the build"+ target_fa[POS - window_coords.start -1])print("number of changes haplo1:")print(count1)print("number of changes haplo2:")print(count2)return haplo_1, haplo_2
make_prediction function
Show the code
def make_prediction(gene, interval, haplo, sequence_one_hot, window =131072): # snp_pos, alt_allelewith tf.device('/GPU:0'): search_gene = gene # gene to predict resized_interval = interval.resize(SEQUENCE_LENGTH) start = resized_interval.start end = resized_interval.end center_pos = start + SEQUENCE_LENGTH//2# figure center position chrom = resized_interval.chr# chromosome#Get exon bin range gene_keys = [gene_key for gene_key in transcriptome.genes.keys() if search_gene in gene_key] gene = transcriptome.genes[gene_keys[0]]#Determine output sequence start seq_out_start = start + seqnn_model.model_strides[0]*seqnn_model.target_crops[0] seq_out_len = seqnn_model.model_strides[0]*seqnn_model.target_lengths[0]#Determine output positions of gene exons gene_slice = gene.output_slice(seq_out_start, seq_out_len, seqnn_model.model_strides[0], False)#Make predictions#y_wtreturn predict_tracks(models, sequence_one_hot)
Get indexes of tracks function
Show the code
# Get indexes of trackdef get_track_idx(tracks): track_idx = [] targets_df['local_index'] = np.arange(len(targets_df))for track in tracks: track_idx.append(targets_df.loc[targets_df['description'] == track]['local_index'].tolist())return track_idx
SEQUENCE_LENGTH =524288n_folds =4#To use only one model fold, change to 'n_folds = 1'rc =True#Average across reverse-complement prediction#Read model parameterswithopen(params_file) as params_open : params = json.load(params_open) params_model = params['model'] params_train = params['train']#Read targetstargets_df = pd.read_csv(targets_file, index_col=0, sep='\t')target_index = targets_df.index#Create local index of strand_pair (relative to sliced targets)if rc : strand_pair = targets_df.strand_pair target_slice_dict = {ix : i for i, ix inenumerate(target_index.values.tolist())} slice_pair = np.array([ target_slice_dict[ix] if ix in target_slice_dict else ix for ix in strand_pair.values.tolist() ], dtype='int32')
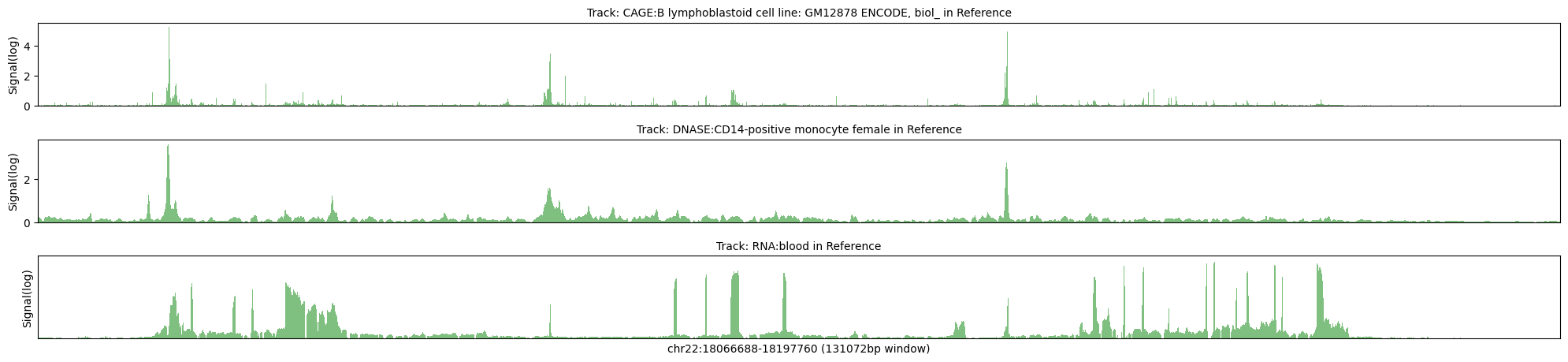
Reference
Max signal for CAGE:B lymphoblastoid cell line: GM12878 ENCODE, biol_ = 38.1346
Max transformed signal for CAGE:B lymphoblastoid cell line: GM12878 ENCODE, biol_ = 5.2904
---
Max signal for DNASE:CD14-positive monocyte female = 11.8511
Max transformed signal for DNASE:CD14-positive monocyte female = 3.6838
---
Max signal for RNA:blood = 2.7051
Max transformed signal for RNA:blood = 1.8895
---
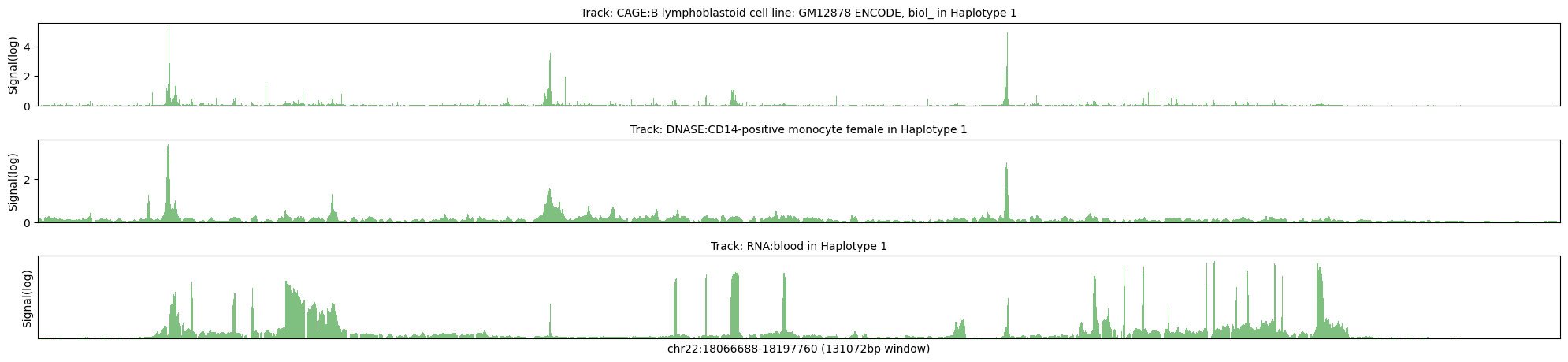
Haplotype 1
Max signal for CAGE:B lymphoblastoid cell line: GM12878 ENCODE, biol_ = 39.2737
Max transformed signal for CAGE:B lymphoblastoid cell line: GM12878 ENCODE, biol_ = 5.3318
---
Max signal for DNASE:CD14-positive monocyte female = 11.8428
Max transformed signal for DNASE:CD14-positive monocyte female = 3.6829
---
Max signal for RNA:blood = 2.689
Max transformed signal for RNA:blood = 1.8832
---
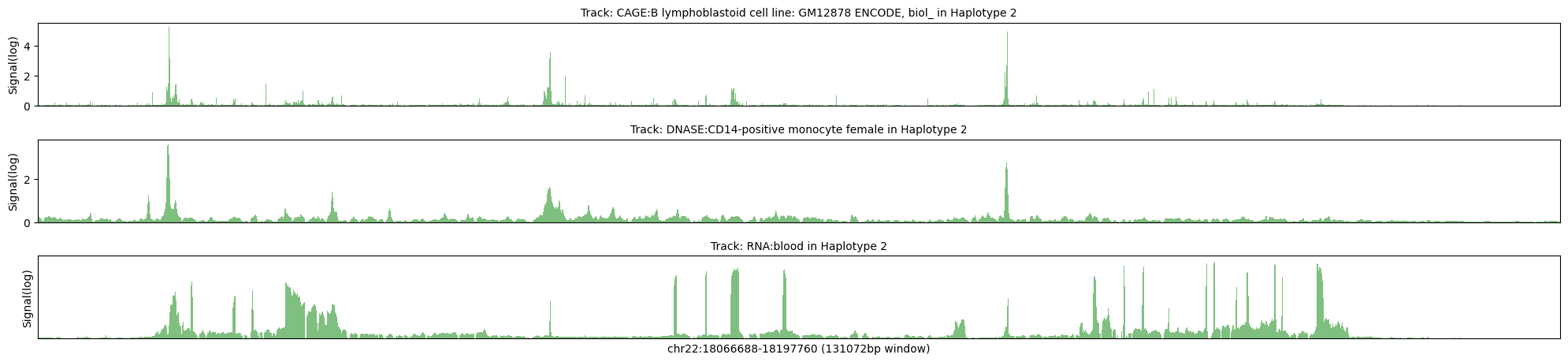
Haplotype 2
Max signal for CAGE:B lymphoblastoid cell line: GM12878 ENCODE, biol_ = 38.5659
Max transformed signal for CAGE:B lymphoblastoid cell line: GM12878 ENCODE, biol_ = 5.3062
---
Max signal for DNASE:CD14-positive monocyte female = 11.8301
Max transformed signal for DNASE:CD14-positive monocyte female = 3.6815
---
Max signal for RNA:blood = 2.7414
Max transformed signal for RNA:blood = 1.9036
---